Tutorial: Adding Demography to COVID Model
Day 5 | Network Modeling for Epidemics
In this tutorial, we will build on our SEIR model for COVID-19 by adding demography (aging, births, and deaths). This will involve learning a few new components of the EpiModel extension API.
Setup
First start by loading EpiModel and clearing your global environment.
library(EpiModel)
rm(list = ls())We will use the extension infection and disease progression modules from Day 4. Like the models with extension modules we ran then, we will keep the functions within one R script and the code to run the model in another script. For the purposes of this tutorial, we will show them bundled together but the linked files are split.
We will start with the initialization of the network object, with the relevant age and disease status attributes, then show the new epidemic models, then the network model estimation, and then epidemic model parameterization and simulation. In practice, we would typically go back and forth many times between all of these components.
Network Initialization
The model will represent age as a continuous nodal attribute. Aging will increment this attribute for everyone by a day (the value of the time step) in a linear fashion, and mortality will be a function of age.
To start, we initialize the range of possible ages (in years) at the outset.
ages <- 0:85Next, we calculate age-specific mortality rates based on one demographic table online. This lists the rates per year per 100,000 persons in the United States by age groups, in the following age bands:
# Rates per 100,000 for age groups: <1, 1-4, 5-9, 10-14, 15-19, 20-24, 25-29,
# 30-34, 35-39, 40-44, 45-49, 50-54, 55-59,
# 60-64, 65-69, 70-74, 75-79, 80-84, 85+
departure_rate <- c(588.45, 24.8, 11.7, 14.55, 47.85, 88.2, 105.65, 127.2,
154.3, 206.5, 309.3, 495.1, 736.85, 1051.15, 1483.45,
2294.15, 3642.95, 6139.4, 13938.3)To convert this to a per-capita daily death rate, we divide by 365,000:
dr_pp_pd <- departure_rate / 1e5 / 365Next we build out a vector of daily death rates by repeating the rate above by the size of the age band. Now we have a rate for each age, with some repeats within age band.
age_spans <- c(1, 4, rep(5, 16), 1)
dr_vec <- rep(dr_pp_pd, times = age_spans)
head(data.frame(ages, dr_vec), 20) ages dr_vec
1 0 1.612192e-05
2 1 6.794521e-07
3 2 6.794521e-07
4 3 6.794521e-07
5 4 6.794521e-07
6 5 3.205479e-07
7 6 3.205479e-07
8 7 3.205479e-07
9 8 3.205479e-07
10 9 3.205479e-07
11 10 3.986301e-07
12 11 3.986301e-07
13 12 3.986301e-07
14 13 3.986301e-07
15 14 3.986301e-07
16 15 1.310959e-06
17 16 1.310959e-06
18 17 1.310959e-06
19 18 1.310959e-06
20 19 1.310959e-06It ends up looking like this visually:
par(mar = c(3,3,2,1), mgp = c(2,1,0), mfrow = c(1,1))
plot(ages, dr_vec, type = "o", xlab = "age", ylab = "Mortality Rate")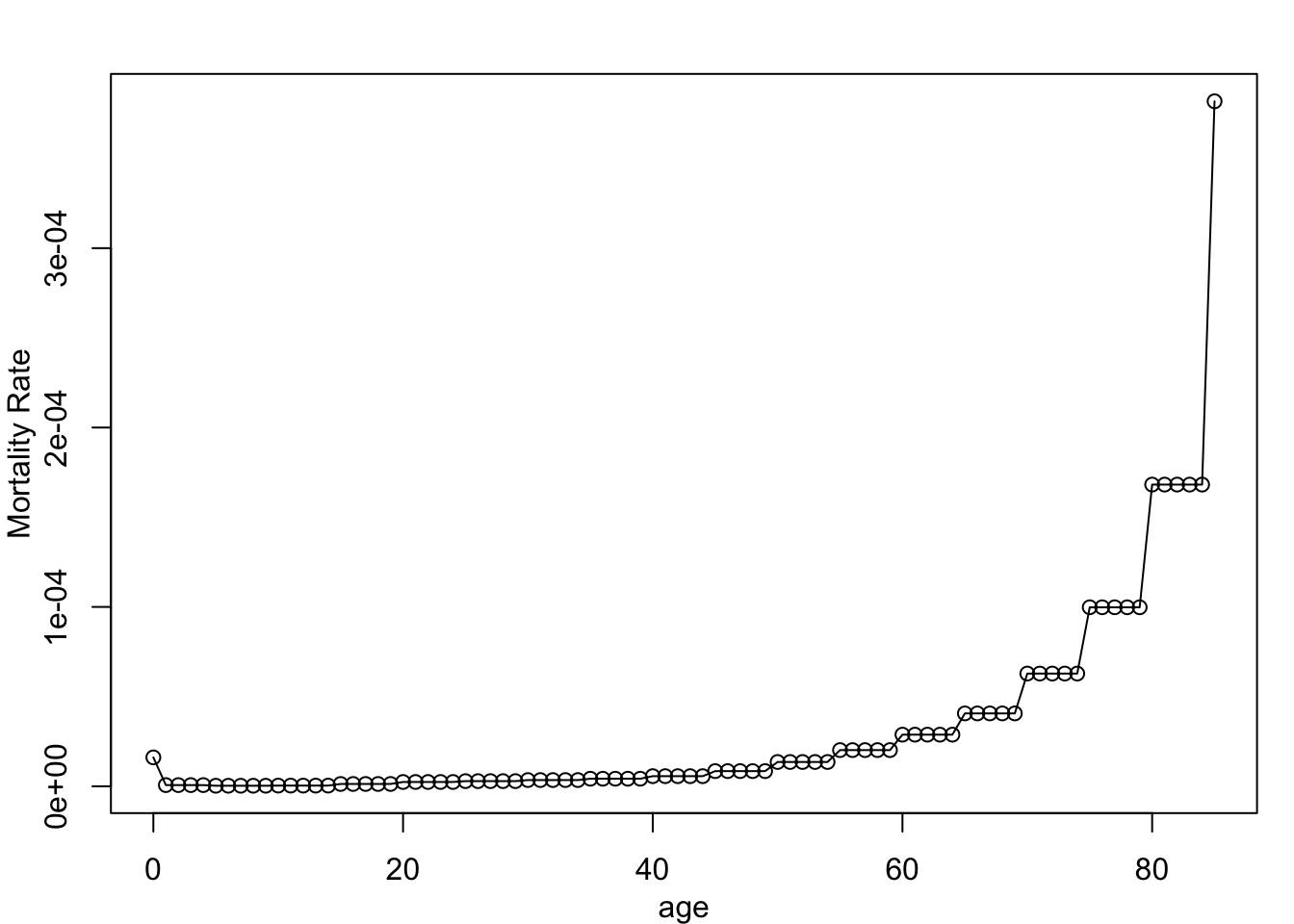
We will now add age and disease status attributes to an initialized network object. We will use a larger population size than last time:
n <- 1000
nw <- network_initialize(n)For each node, we will randomly sample an age from the vector of ages (from 0 to 85), and then set that as a vertex attribute on the network object.
ageVec <- sample(ages, n, replace = TRUE)
nw <- set_vertex_attribute(nw, "age", ageVec)This model will also initialize people into a latent (E state) disease stage.
statusVec <- rep("s", n)
init.latent <- sample(1:n, 50)
statusVec[init.latent] <- "e"
table(statusVec)statusVec
e s
50 950 which(statusVec == "e") [1] 38 43 58 69 100 107 109 152 155 162 184 200 226 236 252 299 353 355 357
[20] 390 441 443 460 464 479 510 515 525 544 551 558 568 578 590 668 675 679 703
[39] 729 761 775 789 797 808 817 886 887 950 957 978Because of that we will manually set up a vector of disease statuses
and set it on the network object as a status attribute.
This is an alternative way to set up disease status, instead of using
the initial conditions in init.net.
nw <- set_vertex_attribute(nw, "status", statusVec)
nw Network attributes:
vertices = 1000
directed = FALSE
hyper = FALSE
loops = FALSE
multiple = FALSE
bipartite = FALSE
total edges= 0
missing edges= 0
non-missing edges= 0
Vertex attribute names:
age status vertex.names
No edge attributesDemographic Modules
With the network initialized, we will now turn to building out the three new extension modules. This set of extensions will add three demographic processes to the model: aging, births, and deaths. The birth and death processes in particular will involve some specific changes to the nodes that must be completed under the EpiModel extension API.
Aging
The aging module performs one simple process: to update the age
attribute (in years) of everyone on the network by a day (or 1/365
years). It does this by getting the age attribute from the
dat object, updating the vector, and then setting it back
on the dat object. The function design follows the same
format as our SEIR modules in Day 4.
aging <- function(dat, at) {
age <- get_attr(dat, "age")
age <- age + 1/365
dat <- set_attr(dat, "age", age)
dat <- set_epi(dat, "meanAge", at, mean(age, na.rm = TRUE))
return(dat)
}We also add a new summary statistic, meanAge, which is
the mean of the age vector for that time step.
Mortality
For the mortality module, we will implement a process of age-specific mortality with an excess risk of death with active infection. The process is more complex than the built-in departures module (which might cover death but other forms of exit from the population). We will step through the function together in the tutorial.
Note that the overall design of the function is similar to any other
EpiModel extension module. The get_ and set_
functions are used to read and write to the dat object. The
individual process of mortality involves querying the death rates by the
age of active nodes, and then drawing from a binomial distribution for
each eligible node.
EpiModel API Rule: For nodes who have died, two
specific nodal attributes must be updated: active and
exitTime. Upon death, the active value of
those nodes turns from 1 to 0. The exitTime is the current
time step, at. These two nodal attributes must be updated
in a death (or other departure) module to tell EpiModel how to
appropriately update the network object at the end of the time step
(which you never have to worry about).
dfunc <- function(dat, at) {
## Attributes
active <- get_attr(dat, "active")
exitTime <- get_attr(dat, "exitTime")
age <- get_attr(dat, "age")
status <- get_attr(dat, "status")
## Parameters
dep.rates <- get_param(dat, "departure.rates")
dep.dis.mult <- get_param(dat, "departure.disease.mult")
## Query alive
idsElig <- which(active == 1)
nElig <- length(idsElig)
## Initialize trackers
nDepts <- 0
idsDepts <- NULL
if (nElig > 0) {
## Calculate age-specific departure rates for each eligible node ##
## Everyone older than 85 gets the final mortality rate
whole_ages_of_elig <- pmin(ceiling(age[idsElig]), 86)
drates_of_elig <- dep.rates[whole_ages_of_elig]
## Multiply departure rates for diseased persons
idsElig.inf <- which(status[idsElig] == "i")
drates_of_elig[idsElig.inf] <- drates_of_elig[idsElig.inf] *
dep.dis.mult
## Simulate departure process
vecDepts <- which(rbinom(nElig, 1, drates_of_elig) == 1)
idsDepts <- idsElig[vecDepts]
nDepts <- length(idsDepts)
## Update nodal attributes
if (nDepts > 0) {
active[idsDepts] <- 0
exitTime[idsDepts] <- at
}
}
## Set updated attributes
dat <- set_attr(dat, "active", active)
dat <- set_attr(dat, "exitTime", exitTime)
## Summary statistics ##
dat <- set_epi(dat, "total.deaths", at, nDepts)
# covid deaths
covid.deaths <- length(intersect(idsDepts, which(status == "i")))
dat <- set_epi(dat, "covid.deaths", at, covid.deaths)
return(dat)
}Note that the overall process of mortality involves flipping a coin for each eligible person in the population, weighted by their age and disease status, and keeping track of who came up as heads (i.e., a 1). We end the function by saving two summary statistics that count the overall deaths and the COVID-related deaths separately.
Births
Finally, we add a new arrivals module to handle births (there may be other forms of arrivals such as in-migration that we would like to handle separately) but not in this model. In terms of the process, births is a simple function of the current population size times a fixed birth rate. This is stochastic at each time step by drawing an non-negative integer from a Poisson distribution.
EpiModel API Rule: If there are any births, then all
the nodal attributes for incoming nodes must be specified. At a minimum,
all EpiModel models must have these 5 attributes: active,
status, infTime, entrTime, and
exitTime. The append_core_attr function
automatically handles the updating of the active,
entrTime, and exitTime attributes, since these
will be uniform across models. Users are responsible for updating
status and infTime with the desired incoming
values, using the append_attr function. Any other nodal
attributes specific to your model, such as age in our
current model, should also be appended using this approach. The
append_attr function allows you to write those standard
values to the end of the attribute vectors, where new nodes are “placed”
in terms of their position on the vector. append_attr can
either take a single value (repeated for all new arrivals) or a vector
of values that may be unique for each arrival.
afunc <- function(dat, at) {
## Parameters ##
n <- get_epi(dat, "num", at - 1)
a.rate <- get_param(dat, "arrival.rate")
## Process ##
nArrivalsExp <- n * a.rate
nArrivals <- rpois(1, nArrivalsExp)
# Update attributes
if (nArrivals > 0) {
dat <- append_core_attr(dat, at = at, n.new = nArrivals)
dat <- append_attr(dat, "status", "s", nArrivals)
dat <- append_attr(dat, "infTime", NA, nArrivals)
dat <- append_attr(dat, "age", 0, nArrivals)
}
## Summary statistics ##
dat <- set_epi(dat, "a.flow", at, nArrivals)
return(dat)
}We close the function by adding a summary statistic for the number of births.
Network Model Estimation
With the new modules defined, we can return to parameterizing the
network model. Recall that we have initialized the network and set the
age and status attributes on the network
above.
nw Network attributes:
vertices = 1000
directed = FALSE
hyper = FALSE
loops = FALSE
multiple = FALSE
bipartite = FALSE
total edges= 0
missing edges= 0
non-missing edges= 0
Vertex attribute names:
age status vertex.names
No edge attributesNext we will parameterize the model starting with the same terms as
our last SEIR model, but adding an absdiff term for age.
This will control the average absolute difference in ages between any
two nodes. With a small target statistic, this will allow for continuous
age homophily (assortative mixing).
formation <- ~edges + degree(0) + absdiff("age")For target statistics, we will start with mean degree, transform that
to the expected edges, and then calculate the number of
isolates (a synonym for degree(0)) as a proportion of
nodes, and absdiff as a function of edges (the target
statistic is the sum of all absolute differences in age across all
edges, so this is the mean difference times the number of edges).
mean_degree <- 2
edges <- mean_degree * (n/2)
avg.abs.age.diff <- 2
isolates <- n * 0.08
absdiff <- edges * avg.abs.age.diff
target.stats <- c(edges, isolates, absdiff)
target.stats[1] 1000 80 2000The dissolution model will use a standard homogeneous dissolution rate, with a death correction here based on the average death rate across the population and a multiplier to account for higher mortality due to COVID. This is not perfect, but this parameter is quite robust to misspecification because the huge differential in average relational duration versus average lifespan (1/mortality rates); notice the minimal change in coefficient with this adjustment.
coef.diss <- dissolution_coefs(~offset(edges), 20, mean(dr_vec)*2)
coef.dissDissolution Coefficients
=======================
Dissolution Model: ~offset(edges)
Target Statistics: 20
Crude Coefficient: 2.944439
Mortality/Exit Rate: 6.31841e-05
Adjusted Coefficient: 2.946969Next we fit the model. You can ignore any warning messages about fitted probabilities that were 0 or 1. These are standard GLM fitting messages. If there is a problem in the model fit, it will emerge in the diagnostics.
est <- netest(nw, formation, target.stats, coef.diss)We will run the model diagnostics by monitoring both the terms in the
model formula and an extension of the degree terms out to degree of 6.
We are using an MCMC control setting here to tweak the simulation
algorithm to increase the “burn-in” for the Markov chain; this burn-in
control is especially helpful in cases of ERGM formulas with
absdiff terms (i.e., those with continuous attributes).
Although the fit is not perfect here (we are on the cusp of where we
also might want to turn off the edges dissolution approximation with
edapprox), it is close enough to move forward for this
tutorial.
dx <- netdx(est, nsims = 10, ncores = 5, nsteps = 500,
nwstats.formula = ~edges + absdiff("age") + degree(0:6),
set.control.tergm = control.simulate.formula.tergm(MCMC.burnin.min = 20000))
Network Diagnostics
-----------------------
- Simulating 10 networks
- Calculating formation statistics
- Calculating duration statistics
- Calculating dissolution statistics
print(dx)EpiModel Network Diagnostics
=======================
Diagnostic Method: Dynamic
Simulations: 10
Time Steps per Sim: 500
Formation Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SD
edges 1000 983.079 -1.692 25.979
absdiff.age 2000 1962.480 -1.876 81.215
degree0 80 89.306 11.632 9.757
degree1 NA 325.059 NA 15.956
degree2 NA 291.031 NA 14.580
degree3 NA 175.243 NA 12.432
degree4 NA 78.899 NA 9.010
degree5 NA 28.833 NA 5.452
degree6 NA 8.692 NA 2.963
Duration Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SD
edges 20 19.998 -0.01 0.099
Dissolution Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SD
edges 0.05 0.05 0.06 0plot(dx)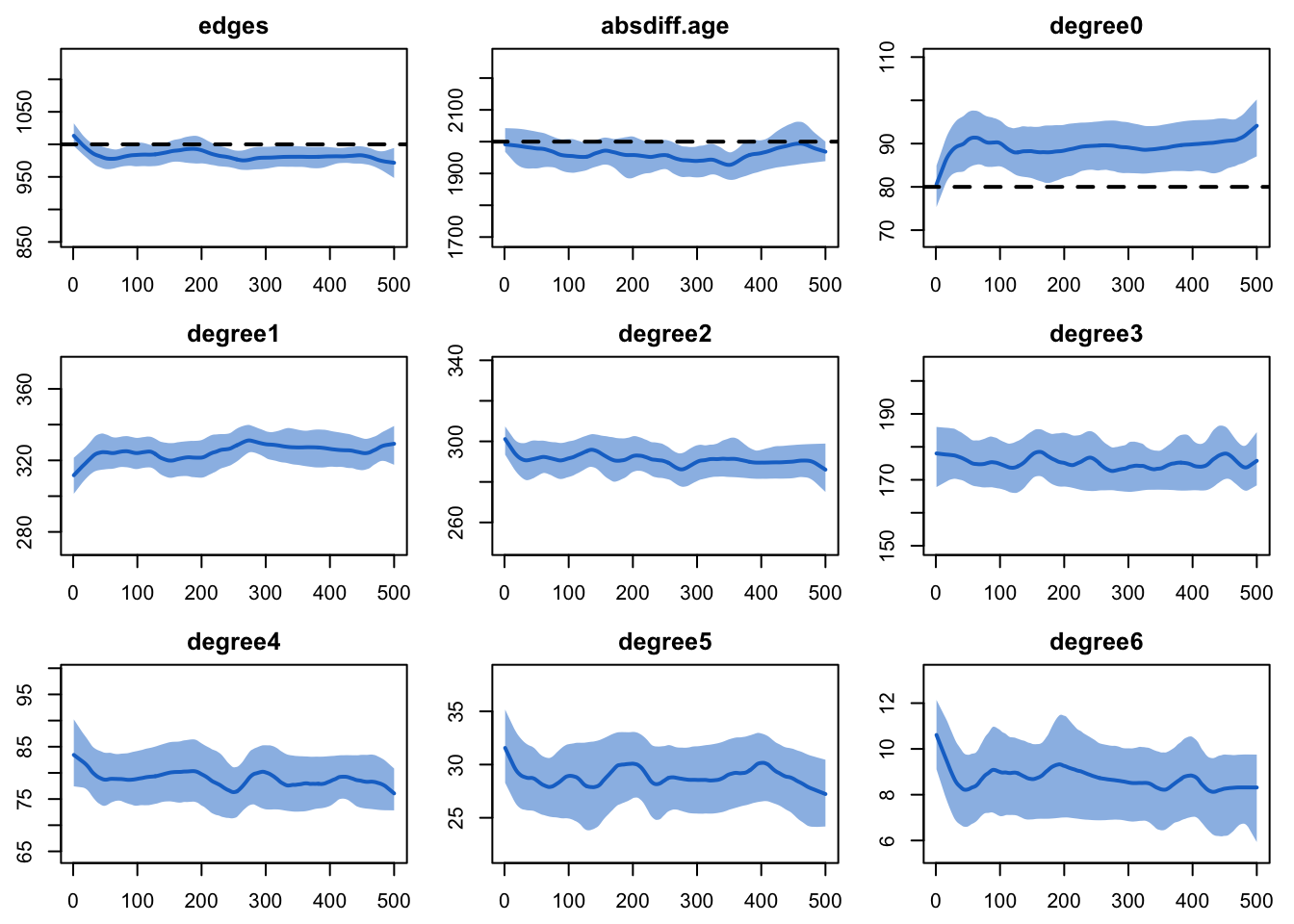
EpiModel Model Simulation
To parameterize the model, we need to add the newly referenced model
parameters to param.net. These include the departure and
arrival rates. Although our past parameters have been scalars (single
values), there is nothing to prevent us from passing vectors (or even
more structured objects like lists or data frames) as parameters. That
is what we do with departure.rates. The
departure.disease.mult is the multiplier on the base death
rate for persons with active infection (persons in the “i” status). We
will assume the arrival rate reflects the average lifespan in days.
param <- param.net(inf.prob = 0.1,
act.rate = 3,
departure.rates = dr_vec,
departure.disease.mult = 100,
arrival.rate = 1/(365*85),
ei.rate = 0.05, ir.rate = 0.05)For initial conditions, we have passed in the number initially in the
latent state at the outset, so init.net just needs to be a
placeholder input.
init <- init.net()Finally, for control settings, we first source in the new function script (this should now contain five extension functions), and include module names and associated functions for each of the five. Note that these five modules will run in addition to the standard modules that handle initialization, network resimulation, and so on. For this model, we definitely want to resimulate the network at each time step, and we will use tergmLite for computational efficiency. We again use a burn-in control setting for TERGM simulations here, plus add some additional network statistics to monitor for diagnostics.
source("d5-s4-COVIDDemog-fx.R")
control <- control.net(type = NULL,
nsims = 5,
ncores = 5,
nsteps = 300,
infection.FUN = infect,
progress.FUN = progress,
aging.FUN = aging,
departures.FUN = dfunc,
arrivals.FUN = afunc,
resimulate.network = TRUE,
tergmLite = TRUE,
set.control.tergm = control.simulate.formula.tergm(MCMC.burnin.min = 10000),
nwstats.formula = ~edges + meandeg + degree(0:2) + absdiff("age"))With all that complete, we now are ready to run netsim.
This should take a minute.
sim <- netsim(est, param, init, control)Post-Simulation Diagnostics
We run the network diagnostics with the netsim
simulations complete to evaluate the potential impact of the epidemic
processes on the network diagnostics. Here we see that the three
targeted statistics are not maintained at their target levels. Part of
this may be due to the general bias in the fit introduced by the edges
dissolution approximation, highlighted in the netdx
diagnostics above. But the change to the edges and
absdiff statistics may be concerning.
plot(sim, type = "formation",
stats = c("edges", "absdiff.age", "degree0"), plots.joined = FALSE)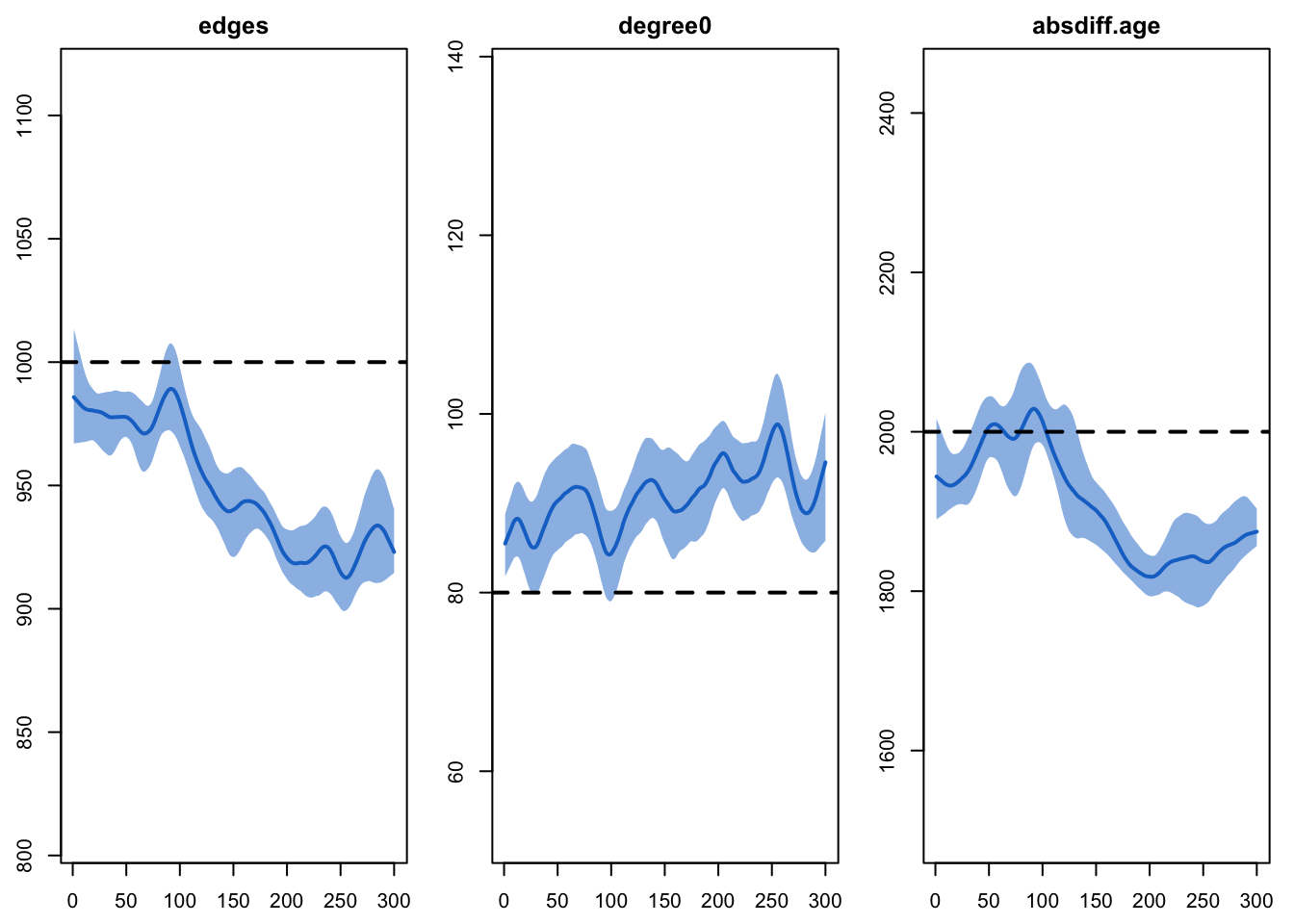
However, it is important to note that the change in edges may be
related to underlying reductions in the population size (as a function
of relatively high COVID-related mortality). To demonstrate this, we
examine the meandeg statistics, which is the per capita
mean degree (i.e., it does not depend on the fluctuations in population
size). Here, the target statistic is relatively stable (perhaps a minor
bias but nothing that will influence the epi outcomes too much).
plot(sim, type = "formation", stats = "meandeg", qnts = 1, ylim = c(1, 3))
abline(h = 2, lty = 2, lwd = 2)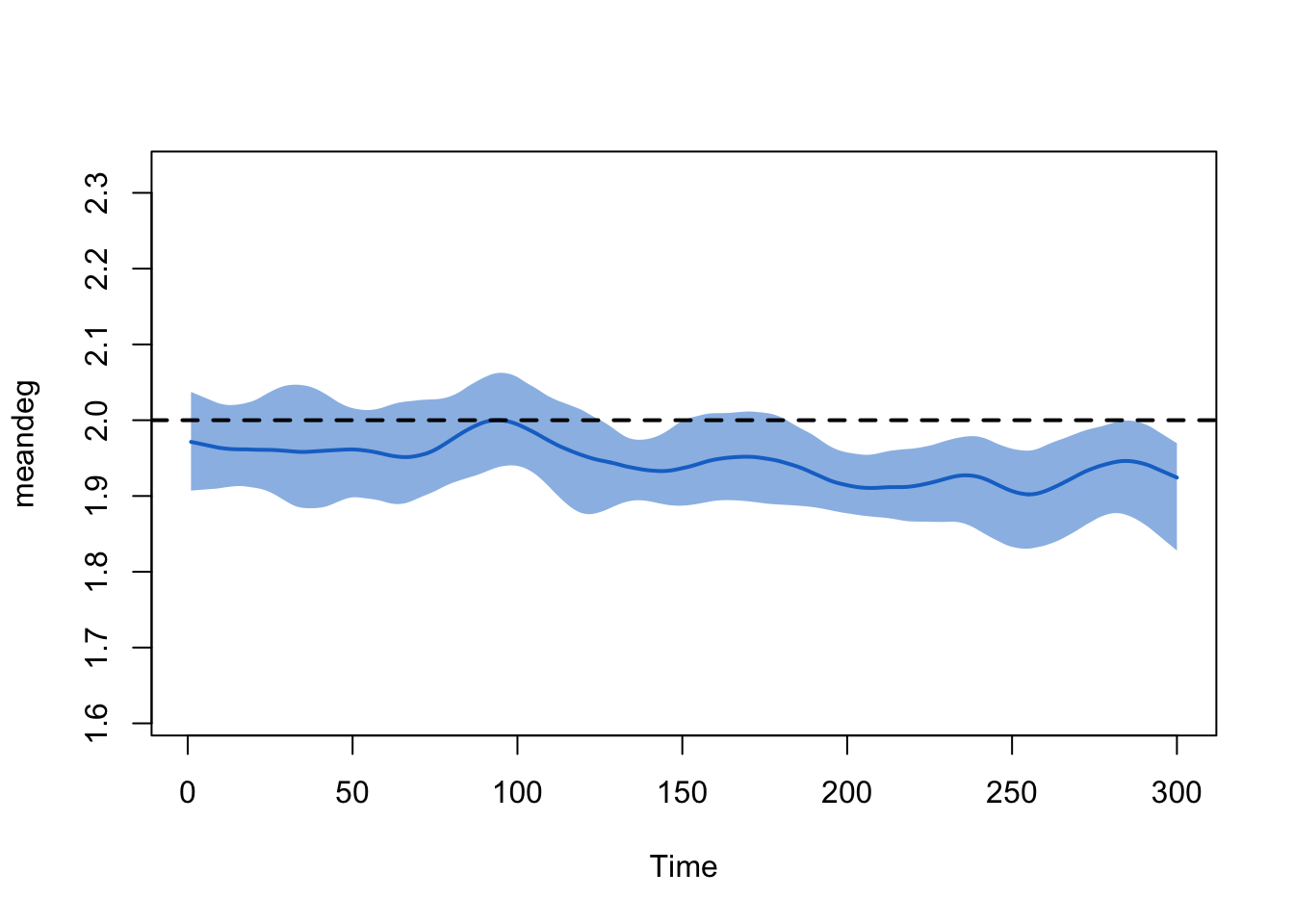
Epidemic Model Analysis
With the epidemic model simulation complete, let’s inspect the
network object. We can see a listing of the modules used, and the model
output variables available for inspection. Because we used
tergmLite, we only have the summary statistic data and not
the individual-level network data.
simEpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type:
No. simulations: 5
No. time steps: 300
No. NW groups: 1
Fixed Parameters
---------------------------
inf.prob = 0.1
act.rate = 3
departure.rates = 1.612192e-05 6.794521e-07 6.794521e-07 6.794521e-07
6.794521e-07 3.205479e-07 3.205479e-07 3.205479e-07 3.205479e-07 3.205479e-07
...
departure.disease.mult = 100
arrival.rate = 3.223207e-05
ei.rate = 0.05
ir.rate = 0.05
groups = 1
Model Functions
-----------------------
initialize.FUN
resim_nets.FUN
infection.FUN
departures.FUN
arrivals.FUN
nwupdate.FUN
prevalence.FUN
verbose.FUN
progress.FUN
aging.FUN
Model Output
-----------------------
Variables: s.num i.num num ei.flow ir.flow e.num r.num
meanAge se.flow total.deaths covid.deaths a.flow
Transmissions: sim1 ... sim5
Formation Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SD
edges 1000 949.230 -5.077 34.946
meandeg NA 1.945 NA 0.053
degree0 80 90.808 13.510 9.930
degree1 NA 318.987 NA 16.066
degree2 NA 284.261 NA 15.872
absdiff.age 2000 1907.789 -4.611 103.735
Dissolution Diagnostics
-----------------------
Not available when:
- `control$tergmLite == TRUE`
- `control$save.network == FALSE`
- dissolution formula is not `~ offset(edges)`Here is a default plot of all the compartment sizes (all variables
ending in .num), with the full quantile range set to 1.
plot(sim, qnts = 1)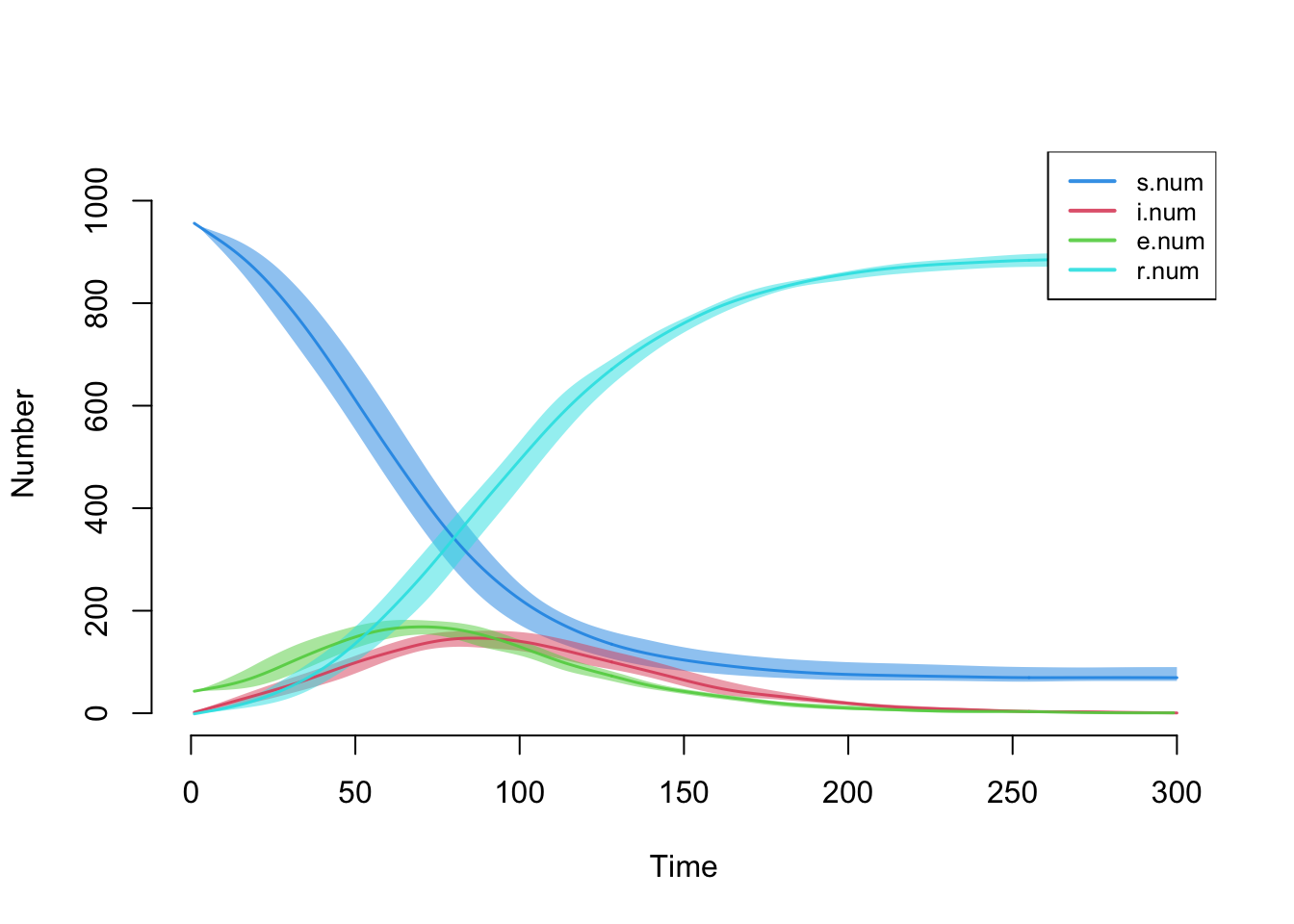
Here is the disease incidence, plotted in two ways:
par(mfrow = c(1,2))
plot(sim, y = "se.flow")
plot(sim, y = "se.flow", mean.smooth = FALSE, qnts = FALSE)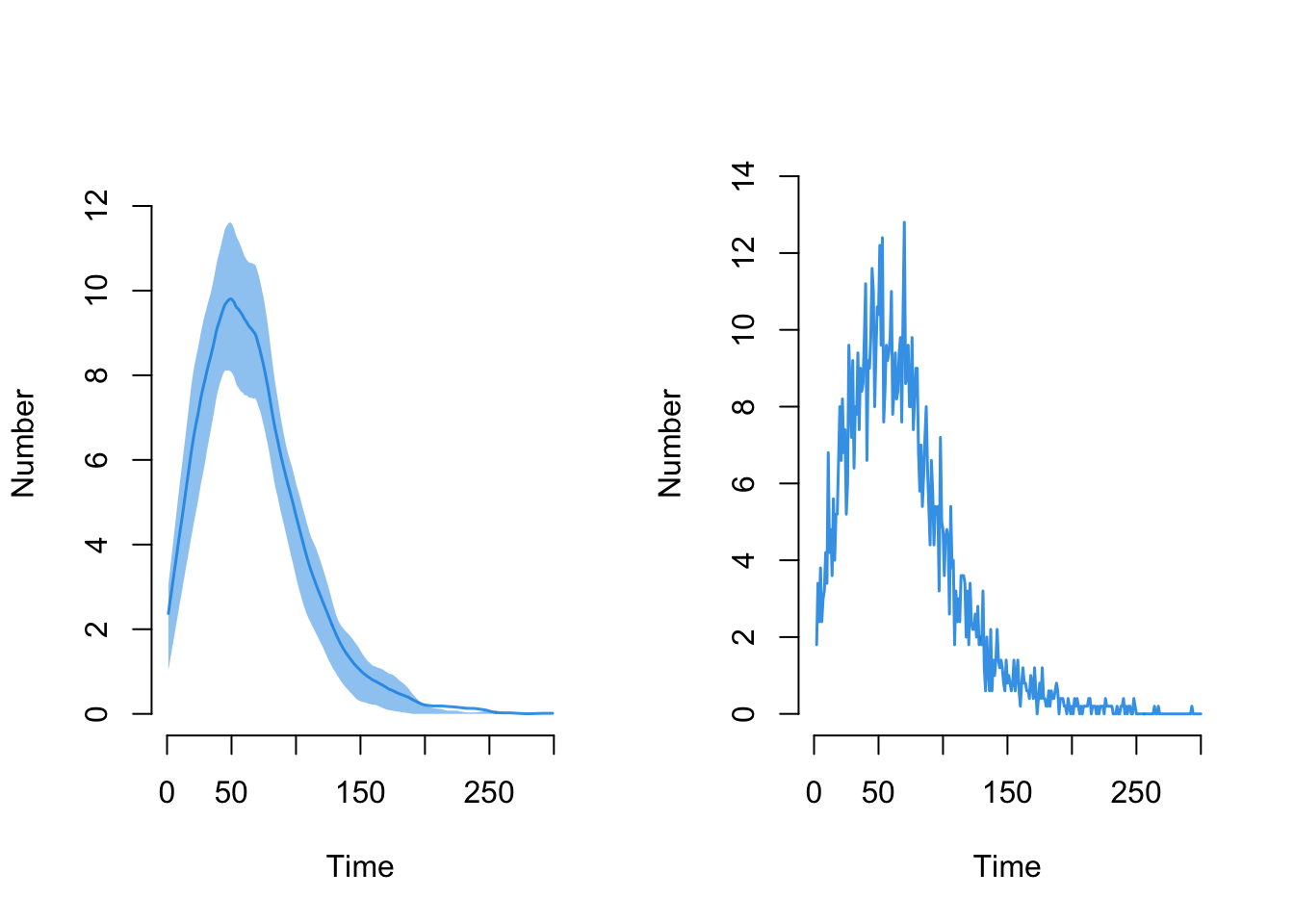
Here is the new mean age summary statistic.
par(mfrow = c(1,1))
plot(sim, y = "meanAge")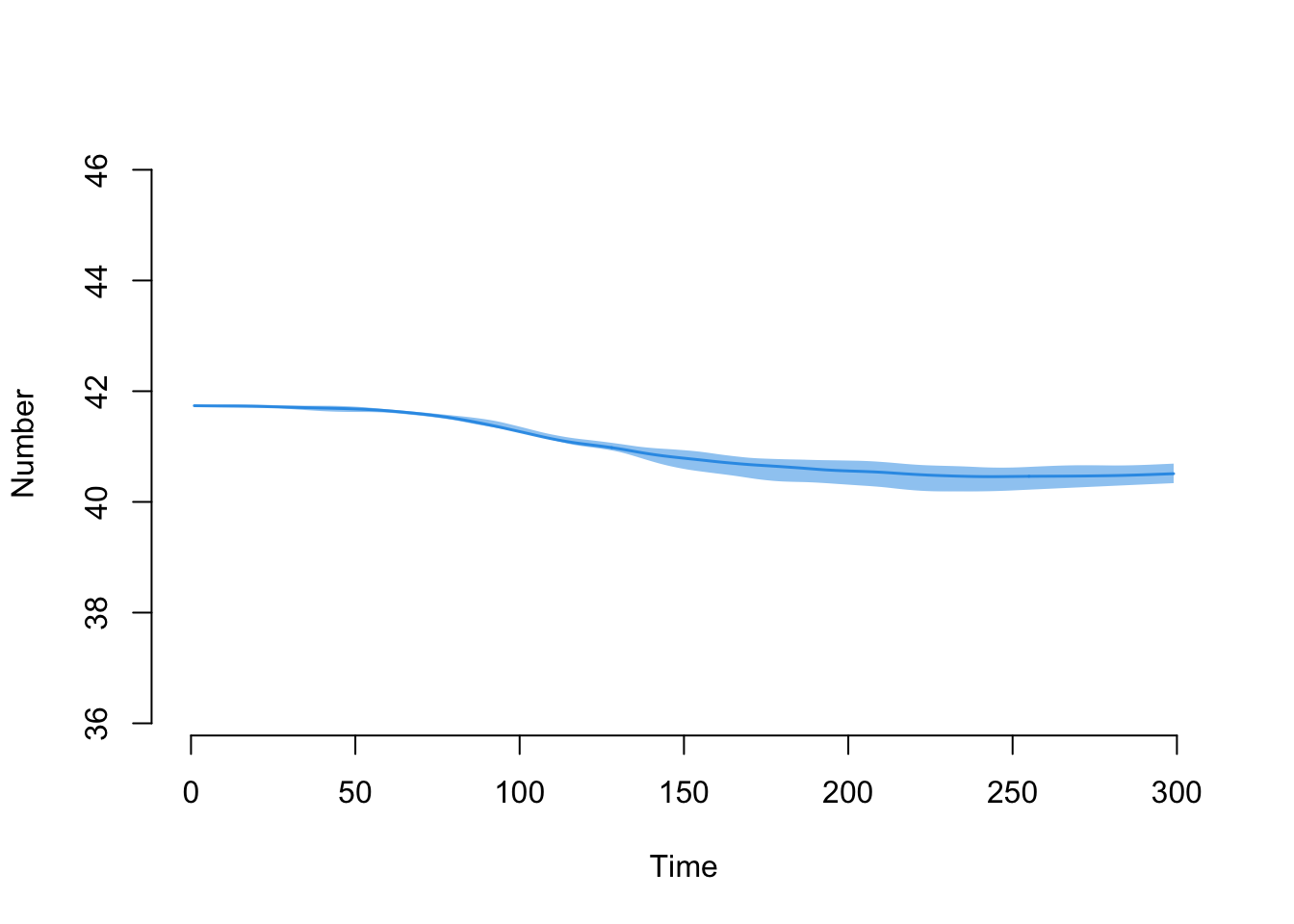
Let’s export the data into a data frame that averaged across the simulations and look at the starting and ending numerical values for mean age:
df <- as.data.frame(sim, out = "mean")
head(df$meanAge)[1] NaN 41.73974 41.73414 41.72882 41.73156 41.73430tail(df$meanAge)[1] 40.49896 40.50170 40.50444 40.50718 40.50992 40.50710Here is the decline in the overall population size over time:
plot(sim, y = "num")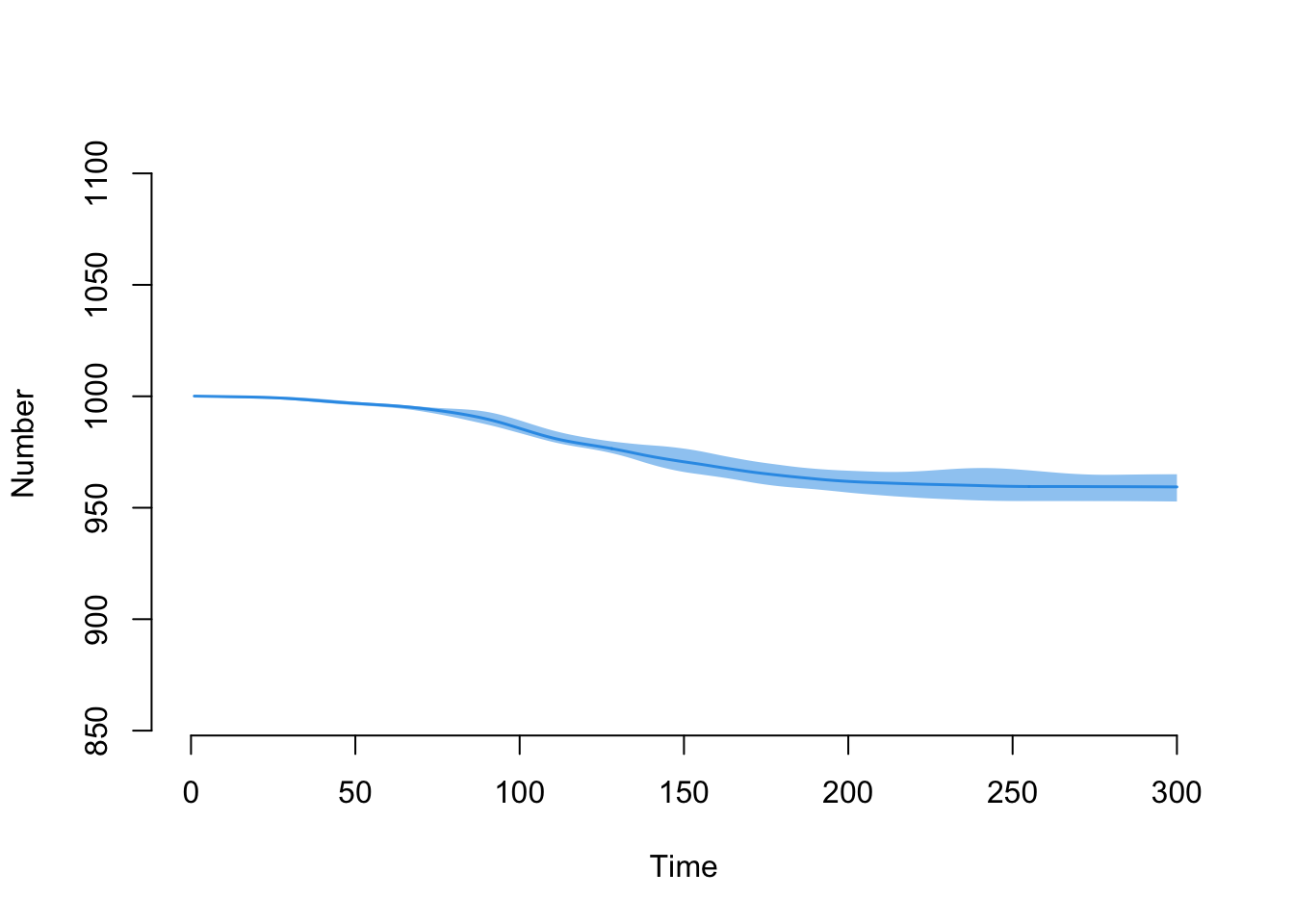
This plot shows the number of new total and COVID-related deaths each day.
plot(sim, y = c("total.deaths", "covid.deaths"), qnts = FALSE, legend = TRUE)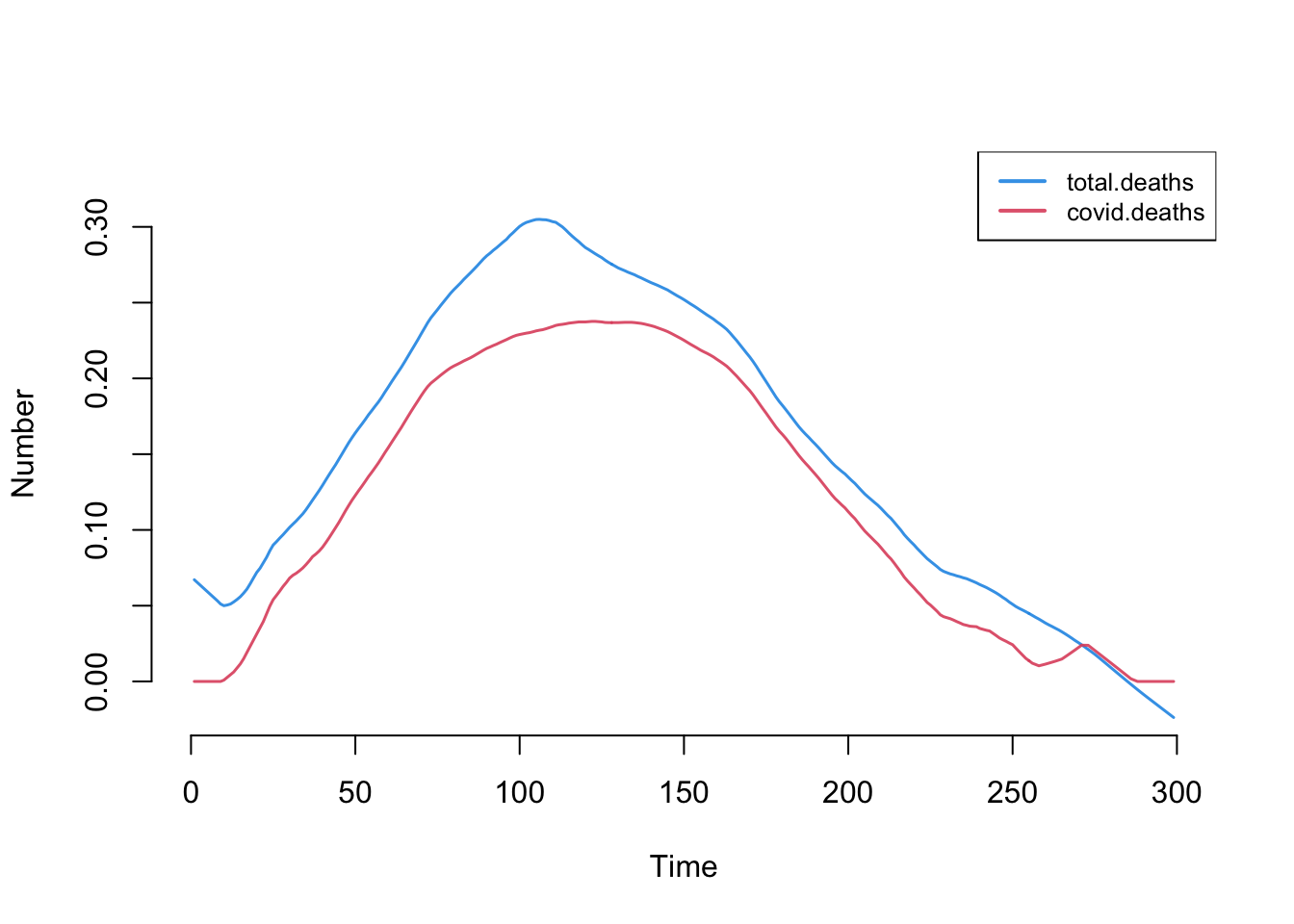
To demonstrate the transmission tree output for this extension model, we can extract the transmission matrix and then plot a phylogram:
tm1 <- get_transmat(sim)
head(tm1, 20) at sus inf transProb actRate finalProb
1 2 230 353 0.1 3 0.271
2 2 420 808 0.1 3 0.271
3 2 224 729 0.1 3 0.271
4 3 455 525 0.1 3 0.271
5 3 371 808 0.1 3 0.271
6 3 175 353 0.1 3 0.271
7 3 413 353 0.1 3 0.271
8 3 841 155 0.1 3 0.271
9 3 985 578 0.1 3 0.271
10 3 858 464 0.1 3 0.271
11 3 588 525 0.1 3 0.271
12 4 123 155 0.1 3 0.271
13 4 84 155 0.1 3 0.271
14 4 769 525 0.1 3 0.271
15 5 537 808 0.1 3 0.271
16 5 166 355 0.1 3 0.271
17 5 448 355 0.1 3 0.271
18 5 953 808 0.1 3 0.271
19 5 895 155 0.1 3 0.271
20 6 512 544 0.1 3 0.271# Plot the phylogram for the first seed
phylo.tm1 <- as.phylo.transmat(tm1)found multiple trees, returning a list of 39phylo objectsplot(phylo.tm1[[1]], show.node.label = TRUE, root.edge = TRUE, cex = 0.75)![]()
Overall, it appears that our new demographic features of the model are performing well!
Last updated: 2022-07-07 with EpiModel v2.3.0